Anxiety Slayer is an Award-Winning anxiety relief podcast with supportive tips, tools, and practices to help you calm anxiety, stress, PTSD, and panic attacks. Celebrating 700 episodes and over 13 million downloads! Visit AnxietySlayer.com to listen in and get our free Anxiety Slayer Starter Course.
…
continue reading
コンテンツは CardioNerds によって提供されます。エピソード、グラフィック、ポッドキャストの説明を含むすべてのポッドキャスト コンテンツは、CardioNerds またはそのポッドキャスト プラットフォーム パートナーによって直接アップロードされ、提供されます。誰かがあなたの著作物をあなたの許可なく使用していると思われる場合は、ここで概説されているプロセスに従うことができますhttps://ja.player.fm/legal。
Cardionerds: A Cardiology Podcastに似ているもの
Seeking a healthier emotional life? It’s time to bring a trained psychologist and mental health expert along for the ride. Dr. Monica Johnson explains the why behind complex emotions, helps you form better relationships, and guides you down the path of self-understanding.
…
continue reading
For the complete newbie or the experienced hypnotist, Brain Software with Mike Mandel is the world's most interesting, educational and fun podcast discussing hypnosis, NLP and strategies for peak performance and self improvement. Mike Mandel is a 6-time award winning hypnotist, immensely popular keynote speaker, stage performer, psychotherapist and NLP trainer. He has been doing hypnosis since he was 12 years old. Mike has an amazing ability to demystify hypnosis, teach core concepts, and te ...
…
continue reading
Practical herbalism from practicing herbalists. Conversations, botanical deep-dives, Q&A with clinical herbalists Katja Swift & Ryn Midura of CommonWealth Holistic Herbalism.
…
continue reading
The Art of Charm is where self-motivated people, just like you, come to learn from the company’s coaches about to how to master human dynamics, relationships, and becoming your best self with the help of Johnny and AJ, the company’s founders. Johnny and AJ bring their 11 years of coaching experience from their famous Bootcamps, where they host clients in Los Angeles from all over the world and they share their stories, best practices and themselves on this weekly podcast. Not only does The A ...
…
continue reading
The Quote of The Day Show is your daily dose of inspiration, featuring the best-of-the-best speakers and prosperity teachers. Each episode spotlights an inspiring quote and 5-10 minute motivational audio clip to help you live a life you love. Featured speakers include Bob Proctor, Lisa Nichols, Tony Robbins, Les Brown, Jim Rohn, and more. Hosted by entrepreneur and money mindset expert Sean Croxton. Follow Sean on IG, Twitter, and FB at @seancroxton. Also, subscribe to his interview podcast, ...
…
continue reading
From the stuff your mother never told you, to the stuff your doctor never learned, On Health features taboo-busting conversations that demystify and de-stigmatize our bodies, all while bridging the gap between conventional medicine and wellness. Join Yale-trained MD & midwife Aviva Romm and her line-up of expert guests as they discuss everything from periods to menopause, sex to reproductive health politics, and motherhood to mental health. Each week, Dr. Romm will be exploring the science a ...
…
continue reading
Do your eyes glaze over when looking at a long list of annual health insurance enrollment options – or maybe while you’re trying to calculate how much you owe the IRS? You might be wondering the same thing we are: Where’s the guidebook for all of this grown-up stuff? Whether opening a bank account, refinancing student loans, or purchasing car insurance (...um, can we just roll the dice without it?), we’re just as confused as you are. Enter: “Grown-Up Stuff: How to Adult” a podcast dedicated ...
…
continue reading
Short guided meditations to calm your anxiety, overcome negative thinking, increase your confidence, and more. Don’t think you have the time, or mental focus, to meditate? Most of these mind-shifting meditations are 10 minutes or less. Soothe your stress away and feel better fast with this award winning guided meditation podcast by Hypnotherapist Chel Hamilton today. For more information visit: https://meditationminis.com (https://meditationminis.com/) Please Note: The meditations presented ...
…
continue reading
Welcome to Almost 30 - a supportive space to fuel your conscious evolution. Join us, LA-based best friends Krista Williams and Lindsey Simcik, for heart-centered, hilarious conversations and real, raw, impactful interviews with brilliant guests. We dive deep into topics like modern spirituality to health and wellness, aliens to entrepreneurship, social justice, and self development. With every episode, our mission is to empower you, expand what you think is possible and, make you laugh - a l ...
…
continue reading
Player FM -ポッドキャストアプリ
Player FMアプリでオフラインにしPlayer FMう!
Player FMアプリでオフラインにしPlayer FMう!

))
405. Case Report: Like Mother, Like Son? Peripartum Cardiomyopathy and Infantile Hypertrophic Cardiomyopathy Lead to a Unifying Diagnosis – Mayo Clinic Arizona
Manage episode 455223435 series 2585945
コンテンツは CardioNerds によって提供されます。エピソード、グラフィック、ポッドキャストの説明を含むすべてのポッドキャスト コンテンツは、CardioNerds またはそのポッドキャスト プラットフォーム パートナーによって直接アップロードされ、提供されます。誰かがあなたの著作物をあなたの許可なく使用していると思われる場合は、ここで概説されているプロセスに従うことができますhttps://ja.player.fm/legal。

CardioNerds (Dr. Dan Ambinder and guest host, Dr. Pooja Prasad) join Dr. Donny Mattia from Phoenix Children’s pediatric cardiology fellowship, Dr. Sri Nayak from the Mayo Clinic – Arizona adult cardiology fellowship, and Dr. Harrison VanDolah from the University of Arizona College of Medicine – Phoenix Med/Peds program for a sunrise hike of Piestewa Peak, followed by some coffee at Berdena’s in Old Town Scottsdale (before the bachelorette parties arrive), then finally a stroll through the Phoenix Desert Botanical Gardens to discuss a thought-provoking case series full of clinical cardiology pearls. Expert commentary is provided by Dr. Tabitha Moe. Episode audio was edited by Dan Ambinder.

They discuss the following case: Cardiology is consulted by the OB team for a 27-year-old female G1, now P1, who has just delivered a healthy baby boy at 34 weeks gestation after going into premature labor. She is experiencing shortness of breath and is found to have a significant past cardiac history, including atrial fibrillation and preexcitation, now with a pacemaker and intracardiac defibrillator. We review the differential diagnosis for peripartum cardiomyopathy (PPCM) and then combine findings from her infant son, who is seen by our pediatric cardiology colleagues and is found to have severe hypertrophic cardiomyopathy (HCM). Genetic testing for both ultimately reveals a LAMP2 mutation consistent with Danon Disease. The case discussion focuses on the differential diagnosis for PPCM, HCM, pearls on Danon Disease and other HCM “phenocopies,” and the importance of good history.
“To study the phenomena of disease without books is to sail an uncharted sea, while to study books without patients is not to go to sea at all.” – Sir William Osler. CardioNerds thank the patients and their loved ones whose stories teach us the Art of Medicine and support our Mission to Democratize Cardiovascular Medicine.
US Cardiology Review is now the official journal of CardioNerds! Submit your manuscript here.
Case Media
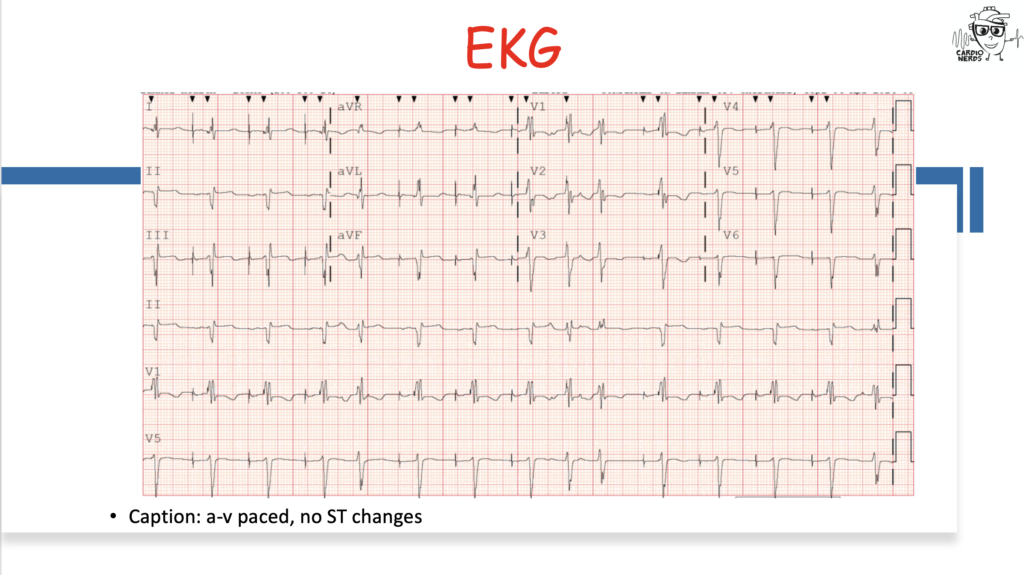
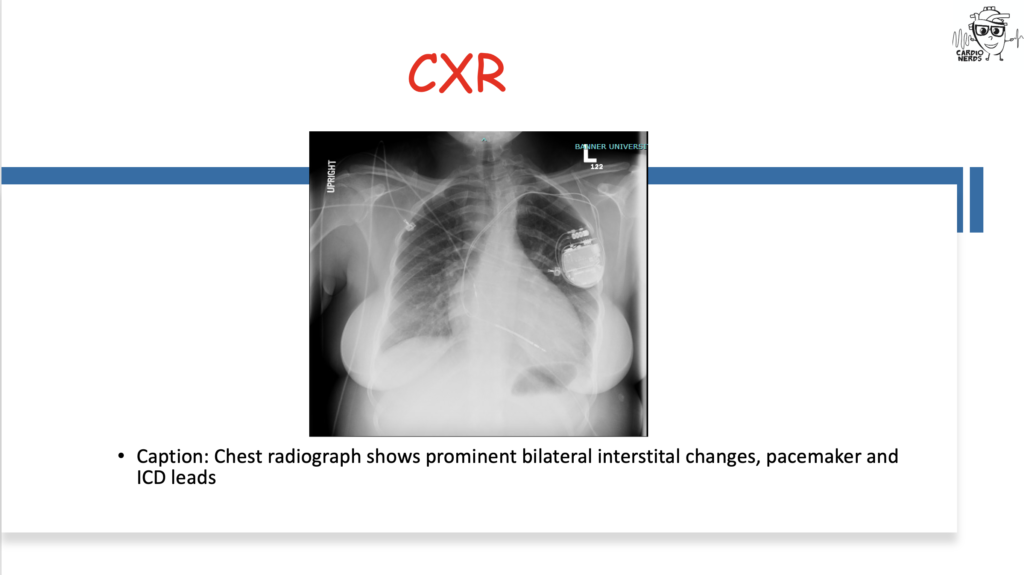
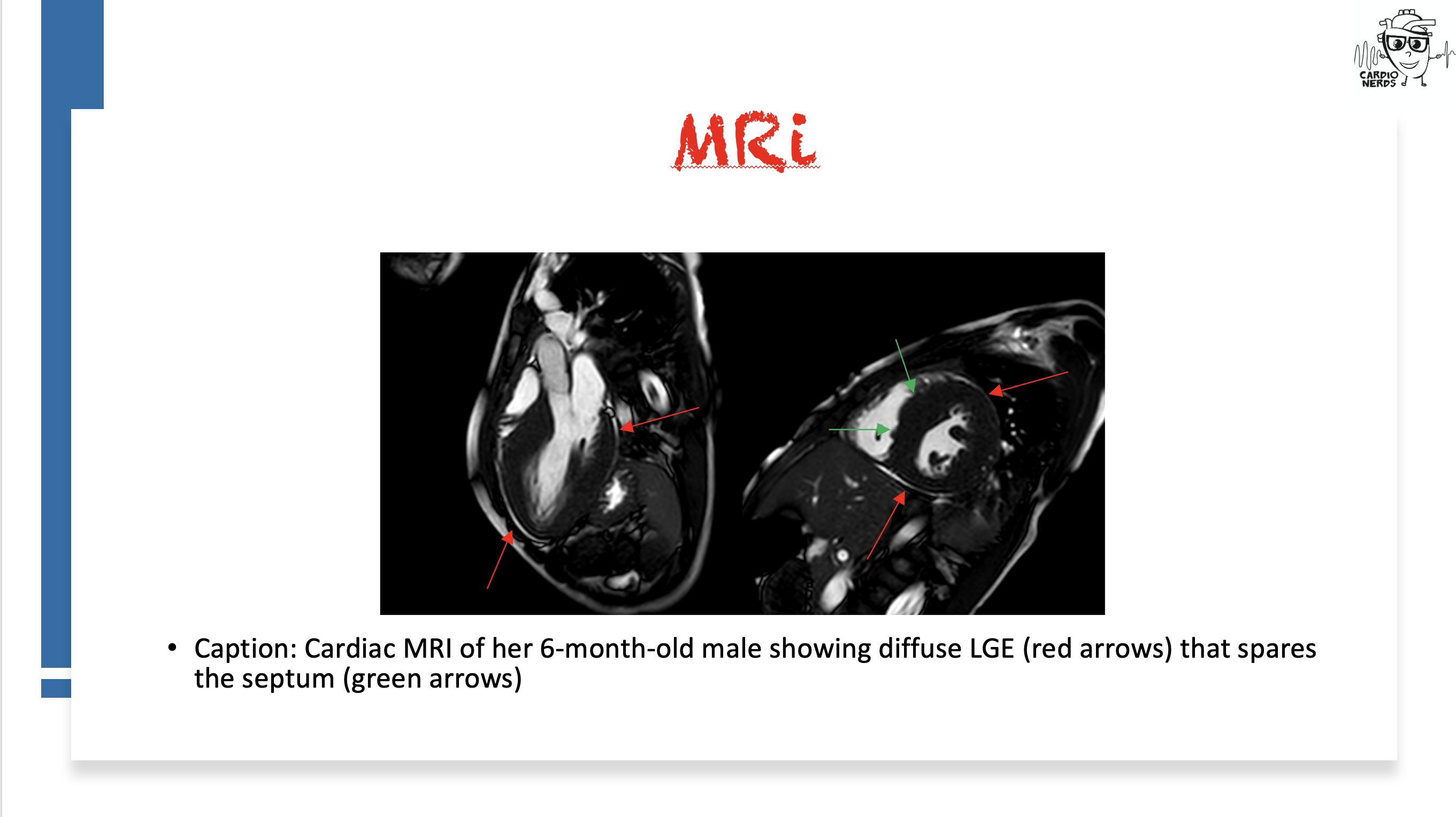
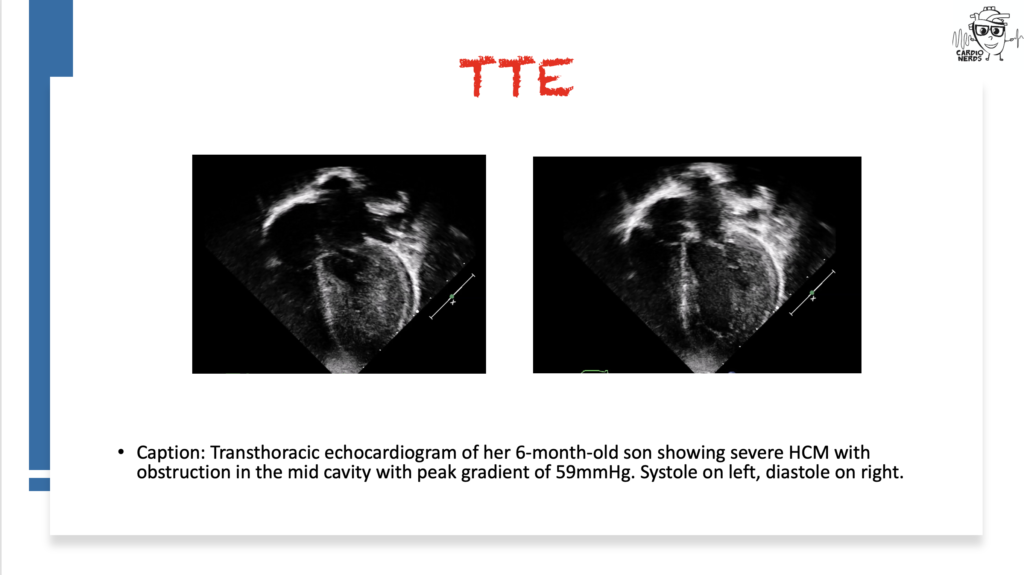
Pearls
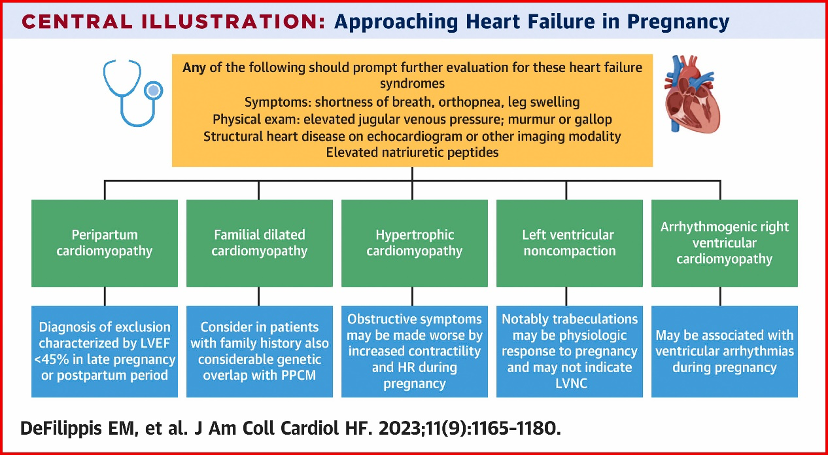
- Peripartum cardiomyopathy is a diagnosis of exclusion – we must exclude other possible etiologies of heart failure!
- Be on the lookout for features of non-sarcomeric HCM – as Dr. Michelle Kittleson said in Episode 166, “LVH plus” states. HCM with preexcitation, heart block, strong family history, or extracardiac symptoms such as peripheral neuropathy, myopathy, or cognitive impairment should be evaluated for infiltrative/inherited cardiomyopathies!
- As an X-linked dominant disorder, Danon disease will present differently in males vs females, with males having much more severe and earlier onset disease with extracardiac features.
- Making the diagnosis for genetic disorders such as Danon disease is important for getting the rest of family members tested as well as the opportunity for specialized treatments such as gene therapy
- Up to 5% of Danon disease cases may be due to copy number variants, which may be missed in genetic testing that does not do targeted deletion/duplication analysis!).
Notes
What is the differential diagnosis for peripartum cardiomyopathy?
- Peripartum cardiomyopathy is a diagnosis of exclusion – we must exclude other possible etiologies of heart failure!
- First, ensure that you are not missing an acute life-threatening etiology of acute decompensated heart failure – pulmonary embolism, amniotic fluid embolism, ACS, and SCAD should all be ruled out.
- Second, a careful history can identify underlying heart disease or risk factors for the development of heart failure, such as substance use, high-risk behaviors that put one at risk for HIV infection, and family history that suggests an inheritable cardiomyopathy.
- Lastly, a careful review of echocardiographic imaging may also identify underlying etiologies that warrant a change in management.
- Diagnosis of peripartum cardiomyopathy is important to consider as within 7 days of onset, patients may be eligible for treatment with bromocriptine – consider referring the patient for enrollment in the ongoing RCT ReBIRTH.
- Check out Cardionerds Episode 113 and the great article linked below for more details on heart failure in pregnancy and postpartum!
What is the differential diagnosis for hypertrophic cardiomyopathy?
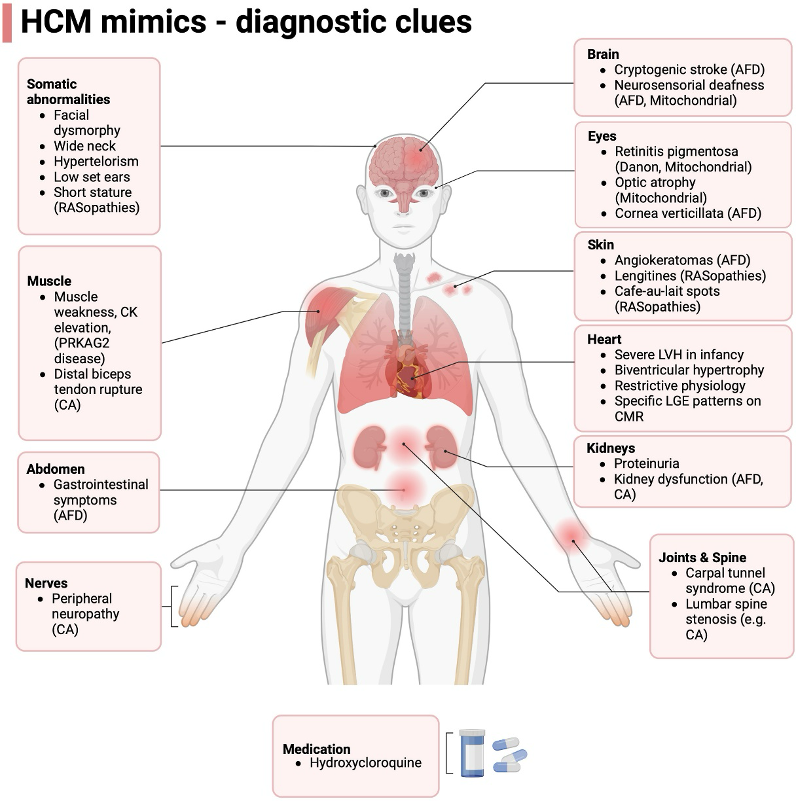
- Though by far the most common differential diagnosis for HCM is simple LVH or athlete’s heart, as Dr. Michelle Kittleson taught us in CardioNerds Episode 166, we should “remain alert for “LVH+” states.”
- It is helpful to think of them in two buckets – sarcomeric mutations (classic HCM) or non-sarcomeric causes (“phenocopies”).
- If you see systemic signs like peripheral neuropathy, renal dysfunction, or skin changes – clues towards a systemic pathology (for adult colleagues, first think amyloidosis; for peds, colleagues, think genetic syndromes such as RASopathies like Noonan syndrome, glycogen, and lysosomal storage diseases like Fabry).
- Additionally, certain additional cardiac findings can point towards a non-sarcomeric HCM – recall way back in CardioNerds Episode 68 when our friends at VCU presented a man in his 60s with a history of WPW/preexcitation and HCM and was found to have a PRKAG2 mutation, which is a similar lysosomal vacuolopathy to Danon disease. Another example was seen in Episode 349 when we saw a patient with HCM and heart block who was found to have Fabry disease.
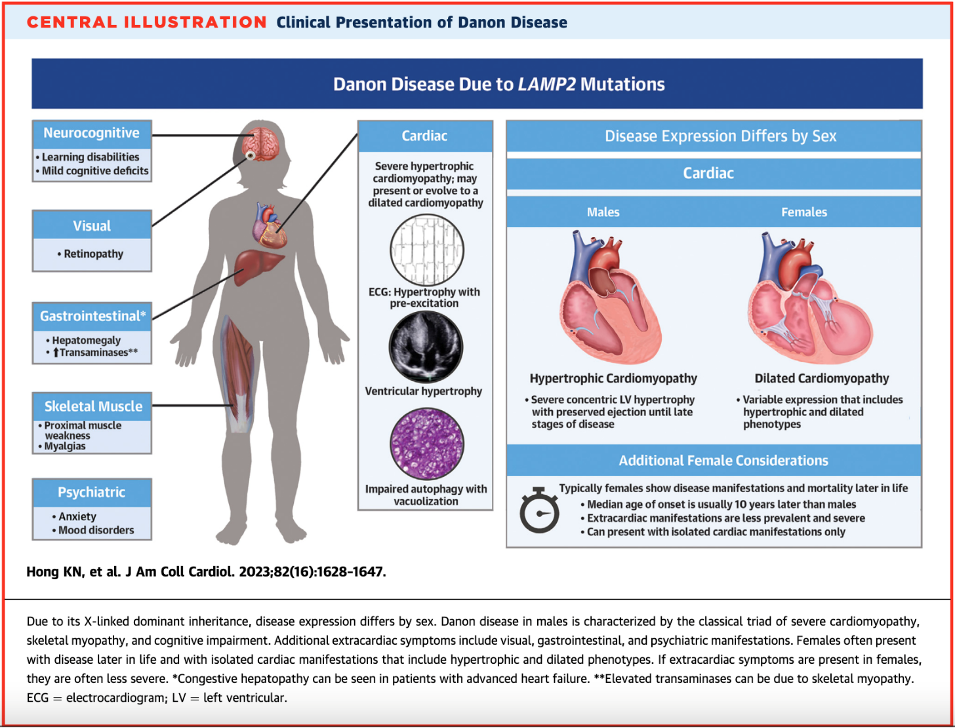
What is Danon disease, and how does it present?
- Danon disease is a rare X-linked dominant genetic disorder due to deficiency in LAMP2, a glycoprotein involved in protecting the lysosome from its roles in endocytosis and autophagy
- When deficiency of LAMP2 occurs, products build up into vacuoles and lead to cardiomyocyte dysfunction and death.
- Interestingly, autophagy disruption is the suspected mechanism of cardiomyopathies from anthracyclines and hydroxychloroquine – Danon disease severity underscores the importance of this process!
- Estimated prevalence of Danon disease in adult patients with HCM is 1-4%, however when both HCM and pre-excitation are present, this rises to 17%.
- It is highly penetrant, meaning most patients with the mutation will show symptoms.
- There are several extracardiac features such as skeletal myopathy, retinopathy, and cognitive impairment – these correlated with areas in the body where LAMP2 is expressed more!
- Classic presentations – remember that X-linked inheritance results in differential expression between males and females!
- Males: young onset with severe LVH/HCM and extracardiac phenotype
- Females: isolated cardiomyopathy (can be either dilated or hypertrophic) with preexcitation arrhythmias with a family history suggesting X-linked dominant transmission (i.e., males more severely affected than females).
- When taking a family history, note that male-to-male transmission (can’t happen since males don’t pass on an X chromosome to their male children) or female-to-offspring transmission (suggests mitochondrial disease) should prompt alternate diagnosis. However, an estimated 1/3 of Danon disease cases are de novo mutations!
- See Episode 300 for a great in-depth overview of the pathophysiology of Danon disease
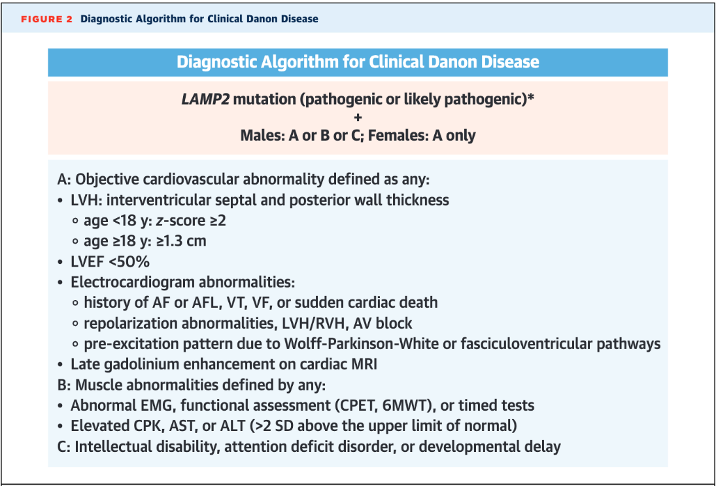
How is Danon disease diagnosed?
- Though there are some proposed characteristic cardiac MRI findings (diffuse LGE sparing the interventricular septum), diagnosis is genetic with a loss-of-function mutation in LAMP2 paired with characteristic cardiac or extracardiac features (see below diagnostic algorithm from Hong et al. JACC 2023)
- LAMP2 is now included in most hypertrophic and dilated CM panels – if found, it is crucial to ensure the patient’s family members also undergo testing and potentially cardiac evaluation! (Note: up to 5% of Danon disease cases may be due to copy number variants, which may be missed in genetic testing that does not do targeted deletion/duplication analysis!)
- Differential Diagnosis
- Sarcomeric HCM – the “classic” HCM, which has numerous genetic causes, all of which affect the sarcomere with age-related penetrance leading to three peaks in age at onset (infancy <1yr, teenage/early adulthood, and mid-adulthood). Progression towards massive LVH and systolic dysfunction is uncommon (<10%) and should raise suspicion of a rare genocopy such as Danon. EKG is usually mostly normal in these patients, unlike in Danon disease, which often has striking abnormalities.
- Pompe disease – lysosome storage disease from mutations in acid alpha-glucosidase leading to lysosomal glycogen accumulation. Autosomal recessive. It can be an infantile form with severe LVH/HCM, and the later forms can have classic skeletal myopathy as well, but usually, these patients have less severe cardiac features.
- RAS-opathies – genetic diseases due to mutations in the RAS/MAPkinase pathway. Classic examples are Noonan syndrome, LEOPARD syndrome, and Costello syndrome. All have classic extracardiac manifestations as well as oftentimes HCM, as well as congenital heart disease such as pulmonary valve stenosis.
- Fabry disease – also an X-linked recessive lysosomal disorder due to alpha-galactosidase A enzyme deficiency; however, it is rarely prominent in childhood and is usually more characterized by extracardiac manifestations. HCM is a cardiac manifestation presenting in the 30s-40s.
- Friedrich ataxia – autosomal recessive multisystem disease due to GAA sequence expansion in the FXN gene that encodes frataxin, a mitochondrial protein, which impairs mitochondrial oxidative phosphorylation. “HCM” is a common disease manifestation in addition to the classic severe neurologic presentation.
- Mitochondrial diseases – heterogenous conditions affecting mitochondrial DNA, transmitted in matrilinear pattern, with cardiac hypertrophy being a classic disease manifestation in addition to preexcitation. These may present with severe cognitive impairment than Danon disease.
- PRKAG2 mutations – cause dysregulation of adenosine monophosphate kinase, culminating in accumulation of vacuoles within glycogen stores. Early-onset cardiac hypertrophy with preexcitation can be similar to Danon disease. There are no extracardiac features and the inheritance pattern is autosomal dominant.
- For a great summary, see Table 2 in Hong K et al. International Consensus on Differential Diagnosis and Management of Patients with Danon disease: JACC state-of-the-art Review. JACC 2023 Oct, 82 (16) 1628-1647.
- For patients who undergo heart transplantation for a genetic/inherited cause, it is crucial to recall their index diagnosis after transplant – they may have extracardiac disease manifestations!
References
- Davis M et al. Peripartum cardiomyopathy: JACC state-of-the-art review. JACC 2020 Jan, 75 (2) 207-221. https://www.jacc.org/doi/10.1016/j.jacc.2019.11.014
- DeFilippis EM et al. Cardio-obstetrics and heart failure: JACC: Heart Failure state-of-the-art review. JACC: HF 2023 Sep, 11(9) 1165-1180. https://www.jacc.org/doi/10.1016/j.jchf.2023.07.009_ga=2.84541658.274573079.1723517623-224546423.1716483762
- Hong K et al. International consensus on differential diagnosis and management of patients with Danon disease: JACC state-of-the-art review. JACC 2023 Oct, 82 (16) 1628- 1647. https://www.jacc.org/doi/abs/10.1016/j.jacc.2023.08.014
- Miliou A et al. Danon cardiomyopathy: specific imaging signs. JACC: Case Rep 2022 Nov 6;4(22):1496-1500. https://www.sciencedirect.com/science/article/pii/S2666084922006015
- Padkins MR, Bell MR. 33-year-old woman with postpartum acute shortness of breath. Mayo Clinic Proceedings 2020;95(9):2000-2004 https://www.mayoclinicproceedings.org/article/S0025-6196(20)30713-8/fulltext
- Rigolli M et al. Cardiac magnetic resonance imaging in Danon disease cardiomyopathy. JACC: Imaging 2021 Feb, 14 (2) 514-516. https://www.jacc.org/doi/10.1016/j.jcmg.2020.08.011
408 つのエピソード
Manage episode 455223435 series 2585945
コンテンツは CardioNerds によって提供されます。エピソード、グラフィック、ポッドキャストの説明を含むすべてのポッドキャスト コンテンツは、CardioNerds またはそのポッドキャスト プラットフォーム パートナーによって直接アップロードされ、提供されます。誰かがあなたの著作物をあなたの許可なく使用していると思われる場合は、ここで概説されているプロセスに従うことができますhttps://ja.player.fm/legal。
CardioNerds (Dr. Dan Ambinder and guest host, Dr. Pooja Prasad) join Dr. Donny Mattia from Phoenix Children’s pediatric cardiology fellowship, Dr. Sri Nayak from the Mayo Clinic – Arizona adult cardiology fellowship, and Dr. Harrison VanDolah from the University of Arizona College of Medicine – Phoenix Med/Peds program for a sunrise hike of Piestewa Peak, followed by some coffee at Berdena’s in Old Town Scottsdale (before the bachelorette parties arrive), then finally a stroll through the Phoenix Desert Botanical Gardens to discuss a thought-provoking case series full of clinical cardiology pearls. Expert commentary is provided by Dr. Tabitha Moe. Episode audio was edited by Dan Ambinder.
They discuss the following case: Cardiology is consulted by the OB team for a 27-year-old female G1, now P1, who has just delivered a healthy baby boy at 34 weeks gestation after going into premature labor. She is experiencing shortness of breath and is found to have a significant past cardiac history, including atrial fibrillation and preexcitation, now with a pacemaker and intracardiac defibrillator. We review the differential diagnosis for peripartum cardiomyopathy (PPCM) and then combine findings from her infant son, who is seen by our pediatric cardiology colleagues and is found to have severe hypertrophic cardiomyopathy (HCM). Genetic testing for both ultimately reveals a LAMP2 mutation consistent with Danon Disease. The case discussion focuses on the differential diagnosis for PPCM, HCM, pearls on Danon Disease and other HCM “phenocopies,” and the importance of good history.
“To study the phenomena of disease without books is to sail an uncharted sea, while to study books without patients is not to go to sea at all.” – Sir William Osler. CardioNerds thank the patients and their loved ones whose stories teach us the Art of Medicine and support our Mission to Democratize Cardiovascular Medicine.
US Cardiology Review is now the official journal of CardioNerds! Submit your manuscript here.
Case Media
Pearls
- Peripartum cardiomyopathy is a diagnosis of exclusion – we must exclude other possible etiologies of heart failure!
- Be on the lookout for features of non-sarcomeric HCM – as Dr. Michelle Kittleson said in Episode 166, “LVH plus” states. HCM with preexcitation, heart block, strong family history, or extracardiac symptoms such as peripheral neuropathy, myopathy, or cognitive impairment should be evaluated for infiltrative/inherited cardiomyopathies!
- As an X-linked dominant disorder, Danon disease will present differently in males vs females, with males having much more severe and earlier onset disease with extracardiac features.
- Making the diagnosis for genetic disorders such as Danon disease is important for getting the rest of family members tested as well as the opportunity for specialized treatments such as gene therapy
- Up to 5% of Danon disease cases may be due to copy number variants, which may be missed in genetic testing that does not do targeted deletion/duplication analysis!).
Notes
What is the differential diagnosis for peripartum cardiomyopathy?
- Peripartum cardiomyopathy is a diagnosis of exclusion – we must exclude other possible etiologies of heart failure!
- First, ensure that you are not missing an acute life-threatening etiology of acute decompensated heart failure – pulmonary embolism, amniotic fluid embolism, ACS, and SCAD should all be ruled out.
- Second, a careful history can identify underlying heart disease or risk factors for the development of heart failure, such as substance use, high-risk behaviors that put one at risk for HIV infection, and family history that suggests an inheritable cardiomyopathy.
- Lastly, a careful review of echocardiographic imaging may also identify underlying etiologies that warrant a change in management.
- Diagnosis of peripartum cardiomyopathy is important to consider as within 7 days of onset, patients may be eligible for treatment with bromocriptine – consider referring the patient for enrollment in the ongoing RCT ReBIRTH.
- Check out Cardionerds Episode 113 and the great article linked below for more details on heart failure in pregnancy and postpartum!
What is the differential diagnosis for hypertrophic cardiomyopathy?
- Though by far the most common differential diagnosis for HCM is simple LVH or athlete’s heart, as Dr. Michelle Kittleson taught us in CardioNerds Episode 166, we should “remain alert for “LVH+” states.”
- It is helpful to think of them in two buckets – sarcomeric mutations (classic HCM) or non-sarcomeric causes (“phenocopies”).
- If you see systemic signs like peripheral neuropathy, renal dysfunction, or skin changes – clues towards a systemic pathology (for adult colleagues, first think amyloidosis; for peds, colleagues, think genetic syndromes such as RASopathies like Noonan syndrome, glycogen, and lysosomal storage diseases like Fabry).
- Additionally, certain additional cardiac findings can point towards a non-sarcomeric HCM – recall way back in CardioNerds Episode 68 when our friends at VCU presented a man in his 60s with a history of WPW/preexcitation and HCM and was found to have a PRKAG2 mutation, which is a similar lysosomal vacuolopathy to Danon disease. Another example was seen in Episode 349 when we saw a patient with HCM and heart block who was found to have Fabry disease.
What is Danon disease, and how does it present?
- Danon disease is a rare X-linked dominant genetic disorder due to deficiency in LAMP2, a glycoprotein involved in protecting the lysosome from its roles in endocytosis and autophagy
- When deficiency of LAMP2 occurs, products build up into vacuoles and lead to cardiomyocyte dysfunction and death.
- Interestingly, autophagy disruption is the suspected mechanism of cardiomyopathies from anthracyclines and hydroxychloroquine – Danon disease severity underscores the importance of this process!
- Estimated prevalence of Danon disease in adult patients with HCM is 1-4%, however when both HCM and pre-excitation are present, this rises to 17%.
- It is highly penetrant, meaning most patients with the mutation will show symptoms.
- There are several extracardiac features such as skeletal myopathy, retinopathy, and cognitive impairment – these correlated with areas in the body where LAMP2 is expressed more!
- Classic presentations – remember that X-linked inheritance results in differential expression between males and females!
- Males: young onset with severe LVH/HCM and extracardiac phenotype
- Females: isolated cardiomyopathy (can be either dilated or hypertrophic) with preexcitation arrhythmias with a family history suggesting X-linked dominant transmission (i.e., males more severely affected than females).
- When taking a family history, note that male-to-male transmission (can’t happen since males don’t pass on an X chromosome to their male children) or female-to-offspring transmission (suggests mitochondrial disease) should prompt alternate diagnosis. However, an estimated 1/3 of Danon disease cases are de novo mutations!
- See Episode 300 for a great in-depth overview of the pathophysiology of Danon disease
How is Danon disease diagnosed?
- Though there are some proposed characteristic cardiac MRI findings (diffuse LGE sparing the interventricular septum), diagnosis is genetic with a loss-of-function mutation in LAMP2 paired with characteristic cardiac or extracardiac features (see below diagnostic algorithm from Hong et al. JACC 2023)
- LAMP2 is now included in most hypertrophic and dilated CM panels – if found, it is crucial to ensure the patient’s family members also undergo testing and potentially cardiac evaluation! (Note: up to 5% of Danon disease cases may be due to copy number variants, which may be missed in genetic testing that does not do targeted deletion/duplication analysis!)
- Differential Diagnosis
- Sarcomeric HCM – the “classic” HCM, which has numerous genetic causes, all of which affect the sarcomere with age-related penetrance leading to three peaks in age at onset (infancy <1yr, teenage/early adulthood, and mid-adulthood). Progression towards massive LVH and systolic dysfunction is uncommon (<10%) and should raise suspicion of a rare genocopy such as Danon. EKG is usually mostly normal in these patients, unlike in Danon disease, which often has striking abnormalities.
- Pompe disease – lysosome storage disease from mutations in acid alpha-glucosidase leading to lysosomal glycogen accumulation. Autosomal recessive. It can be an infantile form with severe LVH/HCM, and the later forms can have classic skeletal myopathy as well, but usually, these patients have less severe cardiac features.
- RAS-opathies – genetic diseases due to mutations in the RAS/MAPkinase pathway. Classic examples are Noonan syndrome, LEOPARD syndrome, and Costello syndrome. All have classic extracardiac manifestations as well as oftentimes HCM, as well as congenital heart disease such as pulmonary valve stenosis.
- Fabry disease – also an X-linked recessive lysosomal disorder due to alpha-galactosidase A enzyme deficiency; however, it is rarely prominent in childhood and is usually more characterized by extracardiac manifestations. HCM is a cardiac manifestation presenting in the 30s-40s.
- Friedrich ataxia – autosomal recessive multisystem disease due to GAA sequence expansion in the FXN gene that encodes frataxin, a mitochondrial protein, which impairs mitochondrial oxidative phosphorylation. “HCM” is a common disease manifestation in addition to the classic severe neurologic presentation.
- Mitochondrial diseases – heterogenous conditions affecting mitochondrial DNA, transmitted in matrilinear pattern, with cardiac hypertrophy being a classic disease manifestation in addition to preexcitation. These may present with severe cognitive impairment than Danon disease.
- PRKAG2 mutations – cause dysregulation of adenosine monophosphate kinase, culminating in accumulation of vacuoles within glycogen stores. Early-onset cardiac hypertrophy with preexcitation can be similar to Danon disease. There are no extracardiac features and the inheritance pattern is autosomal dominant.
- For a great summary, see Table 2 in Hong K et al. International Consensus on Differential Diagnosis and Management of Patients with Danon disease: JACC state-of-the-art Review. JACC 2023 Oct, 82 (16) 1628-1647.
- For patients who undergo heart transplantation for a genetic/inherited cause, it is crucial to recall their index diagnosis after transplant – they may have extracardiac disease manifestations!
References
- Davis M et al. Peripartum cardiomyopathy: JACC state-of-the-art review. JACC 2020 Jan, 75 (2) 207-221. https://www.jacc.org/doi/10.1016/j.jacc.2019.11.014
- DeFilippis EM et al. Cardio-obstetrics and heart failure: JACC: Heart Failure state-of-the-art review. JACC: HF 2023 Sep, 11(9) 1165-1180. https://www.jacc.org/doi/10.1016/j.jchf.2023.07.009_ga=2.84541658.274573079.1723517623-224546423.1716483762
- Hong K et al. International consensus on differential diagnosis and management of patients with Danon disease: JACC state-of-the-art review. JACC 2023 Oct, 82 (16) 1628- 1647. https://www.jacc.org/doi/abs/10.1016/j.jacc.2023.08.014
- Miliou A et al. Danon cardiomyopathy: specific imaging signs. JACC: Case Rep 2022 Nov 6;4(22):1496-1500. https://www.sciencedirect.com/science/article/pii/S2666084922006015
- Padkins MR, Bell MR. 33-year-old woman with postpartum acute shortness of breath. Mayo Clinic Proceedings 2020;95(9):2000-2004 https://www.mayoclinicproceedings.org/article/S0025-6196(20)30713-8/fulltext
- Rigolli M et al. Cardiac magnetic resonance imaging in Danon disease cardiomyopathy. JACC: Imaging 2021 Feb, 14 (2) 514-516. https://www.jacc.org/doi/10.1016/j.jcmg.2020.08.011
408 つのエピソード
Tất cả các tập
×プレーヤーFMへようこそ!
Player FMは今からすぐに楽しめるために高品質のポッドキャストをウェブでスキャンしています。 これは最高のポッドキャストアプリで、Android、iPhone、そしてWebで動作します。 全ての端末で購読を同期するためにサインアップしてください。
Cardionerds: A Cardiology Podcastに似ているもの
Anxiety Slayer is an Award-Winning anxiety relief podcast with supportive tips, tools, and practices to help you calm anxiety, stress, PTSD, and panic attacks. Celebrating 700 episodes and over 13 million downloads! Visit AnxietySlayer.com to listen in and get our free Anxiety Slayer Starter Course.
…
continue reading
Seeking a healthier emotional life? It’s time to bring a trained psychologist and mental health expert along for the ride. Dr. Monica Johnson explains the why behind complex emotions, helps you form better relationships, and guides you down the path of self-understanding.
…
continue reading
For the complete newbie or the experienced hypnotist, Brain Software with Mike Mandel is the world's most interesting, educational and fun podcast discussing hypnosis, NLP and strategies for peak performance and self improvement. Mike Mandel is a 6-time award winning hypnotist, immensely popular keynote speaker, stage performer, psychotherapist and NLP trainer. He has been doing hypnosis since he was 12 years old. Mike has an amazing ability to demystify hypnosis, teach core concepts, and te ...
…
continue reading
Practical herbalism from practicing herbalists. Conversations, botanical deep-dives, Q&A with clinical herbalists Katja Swift & Ryn Midura of CommonWealth Holistic Herbalism.
…
continue reading
The Art of Charm is where self-motivated people, just like you, come to learn from the company’s coaches about to how to master human dynamics, relationships, and becoming your best self with the help of Johnny and AJ, the company’s founders. Johnny and AJ bring their 11 years of coaching experience from their famous Bootcamps, where they host clients in Los Angeles from all over the world and they share their stories, best practices and themselves on this weekly podcast. Not only does The A ...
…
continue reading
The Quote of The Day Show is your daily dose of inspiration, featuring the best-of-the-best speakers and prosperity teachers. Each episode spotlights an inspiring quote and 5-10 minute motivational audio clip to help you live a life you love. Featured speakers include Bob Proctor, Lisa Nichols, Tony Robbins, Les Brown, Jim Rohn, and more. Hosted by entrepreneur and money mindset expert Sean Croxton. Follow Sean on IG, Twitter, and FB at @seancroxton. Also, subscribe to his interview podcast, ...
…
continue reading
From the stuff your mother never told you, to the stuff your doctor never learned, On Health features taboo-busting conversations that demystify and de-stigmatize our bodies, all while bridging the gap between conventional medicine and wellness. Join Yale-trained MD & midwife Aviva Romm and her line-up of expert guests as they discuss everything from periods to menopause, sex to reproductive health politics, and motherhood to mental health. Each week, Dr. Romm will be exploring the science a ...
…
continue reading
Do your eyes glaze over when looking at a long list of annual health insurance enrollment options – or maybe while you’re trying to calculate how much you owe the IRS? You might be wondering the same thing we are: Where’s the guidebook for all of this grown-up stuff? Whether opening a bank account, refinancing student loans, or purchasing car insurance (...um, can we just roll the dice without it?), we’re just as confused as you are. Enter: “Grown-Up Stuff: How to Adult” a podcast dedicated ...
…
continue reading
Short guided meditations to calm your anxiety, overcome negative thinking, increase your confidence, and more. Don’t think you have the time, or mental focus, to meditate? Most of these mind-shifting meditations are 10 minutes or less. Soothe your stress away and feel better fast with this award winning guided meditation podcast by Hypnotherapist Chel Hamilton today. For more information visit: https://meditationminis.com (https://meditationminis.com/) Please Note: The meditations presented ...
…
continue reading
Welcome to Almost 30 - a supportive space to fuel your conscious evolution. Join us, LA-based best friends Krista Williams and Lindsey Simcik, for heart-centered, hilarious conversations and real, raw, impactful interviews with brilliant guests. We dive deep into topics like modern spirituality to health and wellness, aliens to entrepreneurship, social justice, and self development. With every episode, our mission is to empower you, expand what you think is possible and, make you laugh - a l ...
…
continue reading
Player FM -ポッドキャストアプリ
Player FMアプリでオフラインにしPlayer FMう!
Player FMアプリでオフラインにしPlayer FMう!



